Emeriti & Former Faculty
CONTACT
Department of Biological Sciences
Wehr Life Sciences, 109
1428 W. Clybourn St.
Milwaukee, WI 53233
(414) 288-7355


 Dr. Anita ManogaranMarquette University
Dr. Anita ManogaranMarquette UniversityWehr Life Sciences, 313
MilwaukeeWI53201United States of America(414) 288-4580anita.manogaran@marquette.eduLab Websitee-PublicationsB.S., 1996, University of Wisconsin-Milwaukee, Milwaukee, WI
Ph.D., 2003, Marquette University, Milwaukee, WI
Post-doctoral Fellow 2003-2008, University of Illinois at Chicago
Research Assistant Professor, 2008-2010, University of Illinois at Chicago
BIOL 1001 – General Biology 1
BIOL 1410 – Biology of Human Diseases
BIOL 1930 – Foundations in Biological Inquiry
BIOL 2001 – Principles of Biological Investigation
BIOL 2301 – Cell Biology
BIOL 3501 – Neurobiology
BIOL 6005 – Scientific Writing
Protein misfolding is associated with many neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, Huntington’s, and Prion disease. In each of these diseases, a normal protein adopts an alternative but stable conformation that favors aggregation. Although there are genetic determinants that influence the formation of this alternative form, in the vast majority of patients the disease is caused by the sporadic misfolding of proteins. The focus of my lab is to understand the cellular mechanisms that underlie spontaneous protein misfolding associated with disease.
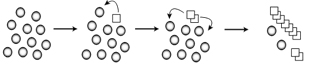
Figure 1. Conversion of a normal protein to the prion form. Normally folded proteins (circles) undergo a rare but stable conformational change into the prion form (squares). The altered conformation can convert more normally folded version of the protein into the prion form.
To begin to understand how these diseases work, our lab focuses on the formation of prions, which are associated with transmissible spongiform encephalopathies, a family of terminal neurological diseases that includes Creutzfeldt-Jakob disease in humans, and “Mad Cow” disease in cattle. In prion disease, a normally folded protein is converted to an alternate conformation that has the ability to further convert the normal protein into the misfolded infectious form (Fig 1). Although much work has focused on looking at infectivity and propagation of prions, much less is known about the initial phases of prion formation since the appearance rates in mammalian systems is approximately 1 in a million. Therefore, the study of a prion in baker’s yeast, [PSI+], has and will continue to be an important model to facilitate our understanding of prion appearance.
To uncover cellular pathways involved in prion formation, our previous work identified several yeast gene deletions that reduced the appearance of the [PSI+] prion. These gene deletions fell into two classes: one class of genes that are involved in the initial misfolding phases of prion appearance, and another class of genes that are important in later steps of prion formation. Interestingly, both classes of genes encode proteins that are involved in actin polymerization and appear to be required to maintain normal vacuolar morphology. Our current work focuses on both genetic approaches and protein biochemistry to investigate how actin networks and the vacuole play an important role in the multistep process of prion appearance.
Claire Radtke (Ph.D. student)
Hannah Buchholz (Ph.D. student)
Adam Knier (Ph.D. student)
Jacqueline Kivila (Ph.D. student)
Dr. Manogaran is currently accepting new Ph.D. students into her lab
Former Students
Doug Lyke, Ph.D. June 2020
Dissertation: Characterization of De Novo Protein Aggregate Formation in S. Cerevisiae

Department of Biological Sciences
Wehr Life Sciences, 109
1428 W. Clybourn St.
Milwaukee, WI 53233
(414) 288-7355
Privacy Policy Legal Disclaimer Non-Discrimination Policy Accessible Technology
© 2026 Marquette University